My understanding is that paired end reads from the Illumina HiSeq/MiSeq platforms looks something like this:
R1:
AAAAAACCCCCC
R2:
GGGGGGTTTTTT
Where the reads found in R2 are the reverse compliment of those found in R1. This does not appear to be the case however, for my sequencing data. If it helps I have a read pair from one of my MiSeq runs below.
R1:
@M01814:86:000000000-A6MU9:1:1101:15397:1339 1:N:0:2
TACTCGCACCTATCCGGCACAGCAACACCATCTGGGGCTGAATCGCAATAGCATCTCTCACTTCCTCCATATCAGATTGCTCAAGGCAAGCACTACGCTGCAGTGCCCTCCACTCCCAATTCCCTGATGCTGGTCGTAACTTGCCACACCA
+
>>AA?BBBBBFFGGG2EEEGFBGHHHGA2FGHBGHF2EE?GHGHHFFEEHDGHEFGF5FEEFBGHGBCB5FHHH5F553@434FF31G11??233B1/1/?333B?3FB?/B24B2/2B2?44?3?23333B223<>@0CB22@2@F0/?/
R2:
@M01814:86:000000000-A6MU9:1:1101:15397:1339 2:N:0:2
TAAGGGGCCTAGAACAGGCACCATACATTCAATTGGCTGTGGCAAGTAACAACCAGCATCAGGGAATGTGGAGTGGAGGGCACTGCAGCGAATTGCTTGCCTTGAACAATCTTATATGGGGGAAGTAGACGAACCAATGTGGAGTCAGCCC
+
>AA>>>ADDAFFGGGGG4FGGGFHFHFHHHFHHHB3B32EFBGGE25FGHHHHACEGG533BAGFFF355331BG1@1>EF1E23F333/>//134B43?F34B3334B334444?443B?/<C/23333////<0/<11111/?01?G0?
Short answer: Normally R1 and R2 are not reverse complements of each other.
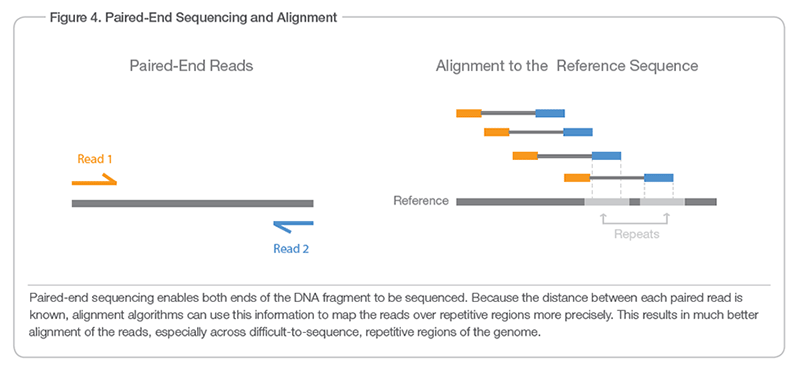
Longer answer: The reverse reads are sequenced in a reversed manner, but the content of the reverse read is not necessarily the reverse complement of the forward read. Most of the time the DNA fragment that you want to sequence is a lot longer than the ~100bp (or up to 300bp depending on the source) that the MiSeq actually can sequence. Therefore the ends of the fragments are sequenced and you only know the sequence of the forward and the reverse read and how far they are apart (inner mate distance if I remember correctly). This graphic from the Illumina website shows that.
Assume you can sequence 10bp and want to sequence a fragment of length 25:
---r1---->
AAAAACCCCCGGGGGTTTTTAAAAA
<----r2---
In this case your inner mate distance is 5 (nr of unseqeunced bases between the reads) and you would get no information on the sequence between the reads (in this example all the Gs). If you analyze a smaller fragment size like this
---r1---->
AAAAACCCCCGGGGG
<----r2---
your reads overlap and you get a negative inner mate distance. Then you get some redundant information as you described, but that normally is not the case.
You can find another helpful article on the manner here.
I hope this helps.
{kind=link}